Last updated: 2024-09-10
Checks: 7 0
Knit directory: My_Project/
This reproducible R Markdown analysis was created with workflowr (version 1.7.1). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20240905) was run prior to running
the code in the R Markdown file. Setting a seed ensures that any results
that rely on randomness, e.g. subsampling or permutations, are
reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 166be7f. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for
the analysis have been committed to Git prior to generating the results
(you can use wflow_publish or
wflow_git_commit). workflowr only checks the R Markdown
file, but you know if there are other scripts or data files that it
depends on. Below is the status of the Git repository when the results
were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Untracked files:
Untracked: .Rapp.history
Untracked: .gitignore
Untracked: Stats.Rmd
Untracked: Stats.html
Untracked: data/.DS_Store
Untracked: data/COADREAD.clin.merged.picked.txt
Untracked: data/COADREAD.rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes_normalized__data.data.txt
Unstaged changes:
Modified: .DS_Store
Modified: analysis/.DS_Store
Deleted: myproject.zip
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were
made to the R Markdown (analysis/EXAMPLE.Rmd) and HTML
(docs/EXAMPLE.html) files. If you’ve configured a remote
Git repository (see ?wflow_git_remote), click on the
hyperlinks in the table below to view the files as they were in that
past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | c3ff74c | oliverdesousa | 2024-09-10 | Build site. |
| Rmd | 5e763e5 | oliverdesousa | 2024-09-10 | Start my new project |
miRseq Analysis:
Analysing miRseq Gene Expression Data from a Colerectal Adenocarcinoma Cohort:
# install.packages("readxl")
# install.packages("readxl")
library(readxl)
library(KODAMA)
library(knitr)Prepare Clinical Data:
# Read in Clinical Data:
coad=read.csv("data/COADREAD.clin.merged.picked.txt",sep="\t",check.names = FALSE)
# Set the first column as row names
rownames(coad) = coad[,1]
# Remove the first column as it is now used as row names
coad = coad[,-1]
coad <- as.data.frame(coad)
# Clean column names: replace dots with dashes & convert to uppercase
colnames(coad) = gsub("\\.", "-", toupper(colnames(coad)))
# Transpose the dataframe so that rows become columns and vice versa
coad = t(coad) Prepare miRNA-seq expression data:
# Read RNA-seq expression data:
r = read.csv("data/COADREAD.rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes_normalized__data.data.txt", sep = "\t", check.names = FALSE, row.names = 1)
# Remove the first row:
r = r[-1,]
# Convert expression data to numeric matrix format
temp = matrix(as.numeric(as.matrix(r)), ncol=ncol(r))
# Assign original column names to the matrix
colnames(temp) = colnames(r)
# Assign original row names to the matrix
rownames(temp) = rownames(r)
RNA = temp
# Transpose the matrix so that genes are rows and samples are columns
RNA = t(RNA) Extract patient and tissue information from column names:
tcgaID = list()
# Extract sample ID
tcgaID$sample.ID <- substr(colnames(r), 1, 16)
# Extract patient ID
tcgaID$patient <- substr(colnames(r), 1, 12)
# Extract tissue type
tcgaID$tissue <- substr(colnames(r), 14, 16)
tcgaID = as.data.frame(tcgaID) Select Primary Solid Tumor tissue data (“01A”):
tcgaID.sel = tcgaID[tcgaID$tissue == "01A", ]
# Subset the RNA expression data to match selected samples
RNA.sel = RNA[tcgaID$tissue == "01A", ]Intersect patient IDs between clinical and RNA data:
sel = intersect(tcgaID.sel$patient, rownames(coad))
# Subset the clinical data to include only selected patients:
coad.sel = coad[sel, ]
# Assign patient IDs as row names to the RNA data:
rownames(RNA.sel) = tcgaID.sel$patient
# Subset the RNA data to include only selected patients
RNA.sel = RNA.sel[sel, ]Prepare labels for pathology stages:
Classify stages
t1,t2, &t3as “low”Classify stages
t4,t4a, &t4bas “high”Convert any
tisstages toNA
labels = coad.sel[, "pathology_T_stage"]
labels[labels %in% c("t1", "t2", "t3")] = "low"
labels[labels %in% c("t4", "t4a", "t4b")] = "high"
labels[labels == "tis"] = NALog Transform the expression data for our selected gene
CXCL2:
CXCL2 <- log(1 + RNA.sel[, "CXCL2|2920"])Boxplot to visualize the distribution of log transformed gene expression by pathology stage:
boxplot(CXCL2 ~ labels, main = "Expression of CXCL2 by Pathology Stage",
xlab = "Pathology Stage",
ylab = "Log Transformed Expression",
col = c("lightyellow", "sandybrown")) 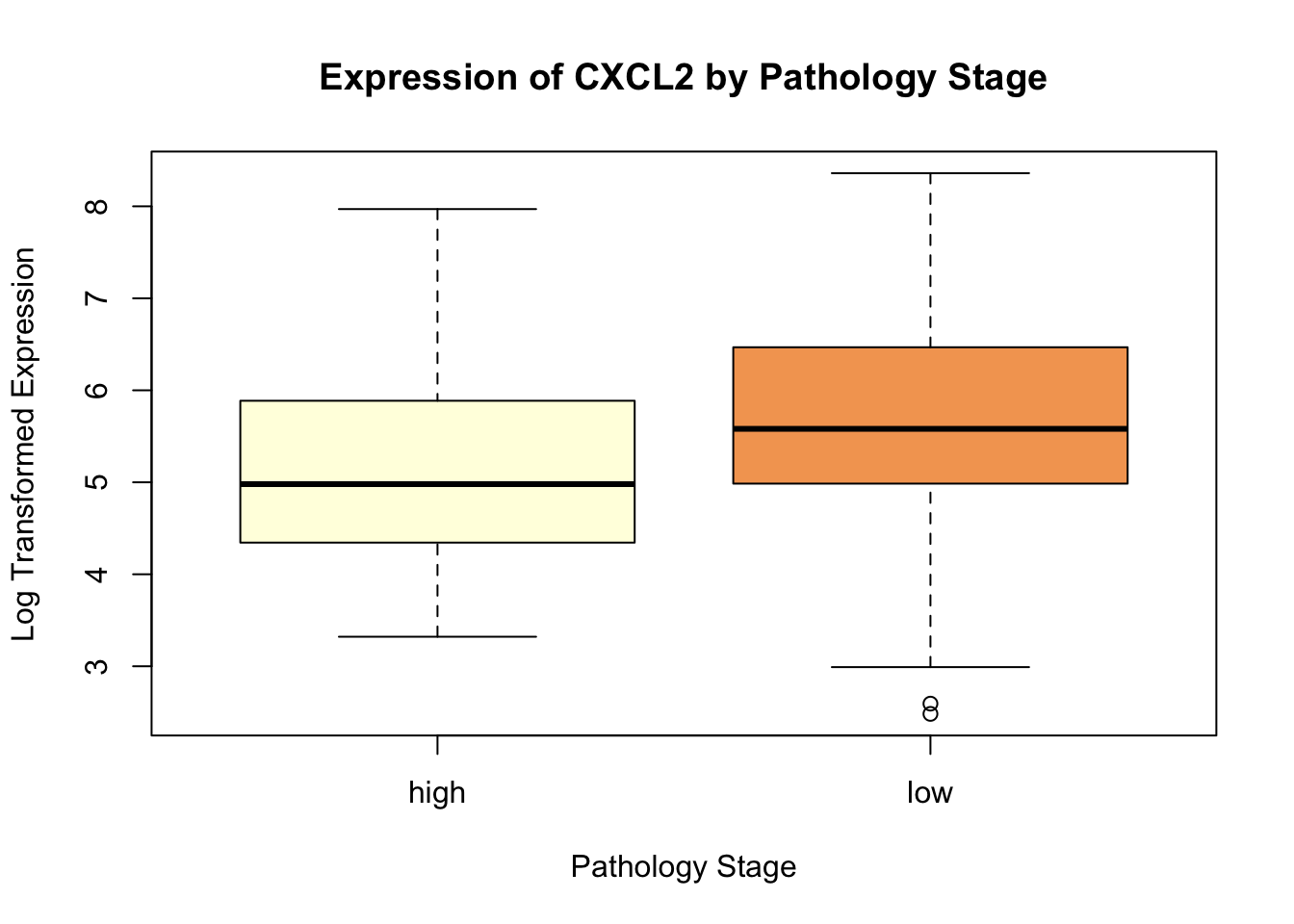
| Version | Author | Date |
|---|---|---|
| c3ff74c | oliverdesousa | 2024-09-10 |
Perform Wilcoxon rank-sum test to compare gene expression between “low” and “high” stages:
CXCL2_W <- wilcox.test(CXCL2 ~ labels)
CXCL2_W
Wilcoxon rank sum test with continuity correction
data: CXCL2 by labels
W = 5357, p-value = 0.0007265
alternative hypothesis: true location shift is not equal to 0Now, we log transform expression data for three other genes:
CXCL3 <- log(1 + RNA.sel[, "CXCL3|2921"])
GPX2 <- log(1 + RNA.sel[, "GPX2|2877"])
LCN2 <- log(1 + RNA.sel[, "LCN2|3934"])Visualize the log transformed gene expression by pathology stage for all four genes:
par(mfrow = c(1, 4))
ylim_range <- c(2, 12)
# CXCL2 boxplot:
boxplot(CXCL2 ~ labels, main = "CXCL2",
xlab = "Pathology Stage",
ylab = "Log Transformed Expression",
col = c("lightyellow", "sandybrown"),
ylim = ylim_range)
# GPX2 boxplot:
boxplot(GPX2~ labels,
main = "GPX2",
xlab = "Pathology Stage",
ylab = "Log Transformed Expression",
col = c("lightblue", "lightcoral"),
ylim = ylim_range) # Specify colors for each box
# CXCL3 boxplot:
boxplot(CXCL3 ~ labels,
main = "CXCL3",
xlab = "Pathology Stage",
ylab = "Log Transformed Expression",
col = c("lightgreen", "lightgoldenrod"),
ylim = ylim_range) # Specify colors for each box
# LCN2 boxplot:
boxplot(LCN2 ~ labels,
main = "LCN2",
xlab = "Pathology Stage",
ylab = "Log Transformed Expression",
col = c("lightpink", "lightsteelblue"),
ylim = ylim_range) # Specify colors for each box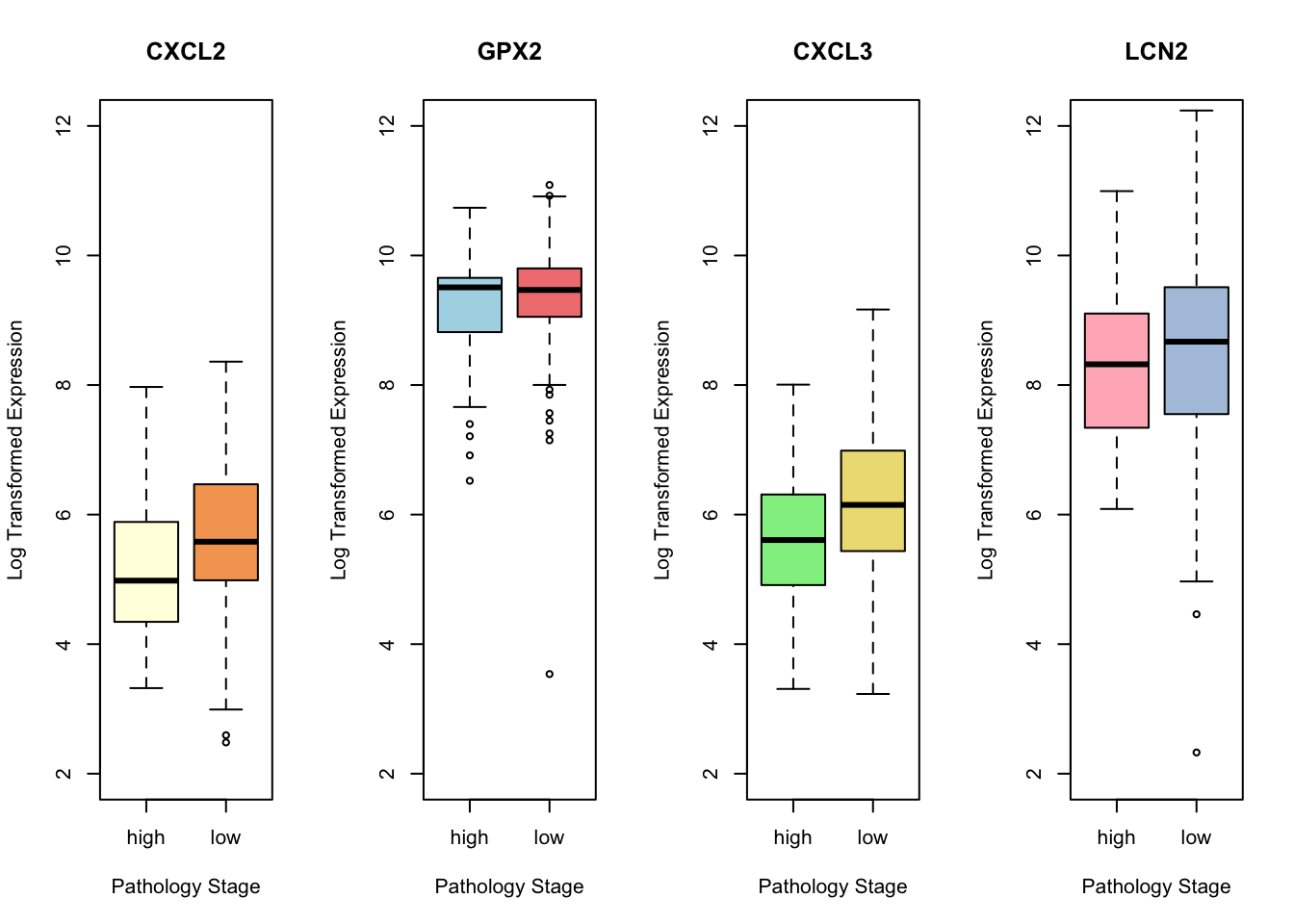
| Version | Author | Date |
|---|---|---|
| c3ff74c | oliverdesousa | 2024-09-10 |
par(mfrow = c(1, 1))Perform Wilcoxon rank-sum test to compare gene expression between “low” and “high” stages for the three new genes:
# LCN2 Gene:
LCN2_W <- wilcox.test(LCN2~ labels)
# CXCL3 Gene:
CXCL3_W <- wilcox.test(CXCL3 ~ labels)
# GPX2 Gene:
GPX2_W <- wilcox.test(GPX2 ~ labels)Now lets compare their output from the Wilcoxon rank-sum test:
results <- data.frame(
Gene = c("LCN2", "CXCL3", "GPX2", "CXCL2"),
Test_Statistic = c(LCN2_W$statistic, CXCL3_W$statistic, GPX2_W$statistic, CXCL2_W$statistic),
P_Value = c(LCN2_W$p.value, CXCL3_W$p.value, GPX2_W$p.value, CXCL2_W$p.value),
stringsAsFactors = FALSE
)
kable(results, digits = 4)| Gene | Test_Statistic | P_Value |
|---|---|---|
| LCN2 | 6916 | 0.2666 |
| CXCL3 | 5119 | 0.0002 |
| GPX2 | 7083 | 0.3854 |
| CXCL2 | 5357 | 0.0007 |
Interpretation:
CXCL2 (0.0007) and CXCL3 (0.0002) showed significant differences in gene expression between high and low pathology stages, with p-values < 0.05.
This result suggests that these genes could be important in distinguishing between different stages of the disease and warrants further investigation into their roles and potential as biomarkers or therapeutic targets.
sessionInfo()R version 4.4.1 (2024-06-14)
Platform: aarch64-apple-darwin20
Running under: macOS Sonoma 14.6.1
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
time zone: Africa/Johannesburg
tzcode source: internal
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] knitr_1.48 KODAMA_3.1 umap_0.2.10.0 Rtsne_0.17 minerva_1.5.10
[6] readxl_1.4.3
loaded via a namespace (and not attached):
[1] Matrix_1.7-0 jsonlite_1.8.8 highr_0.11 compiler_4.4.1
[5] promises_1.3.0 Rcpp_1.0.13 stringr_1.5.1 git2r_0.33.0
[9] parallel_4.4.1 later_1.3.2 jquerylib_0.1.4 png_0.1-8
[13] yaml_2.3.10 fastmap_1.2.0 reticulate_1.38.0 lattice_0.22-6
[17] R6_2.5.1 workflowr_1.7.1 tibble_3.2.1 openssl_2.2.1
[21] rprojroot_2.0.4 bslib_0.8.0 pillar_1.9.0 rlang_1.1.4
[25] utf8_1.2.4 cachem_1.1.0 stringi_1.8.4 httpuv_1.6.15
[29] xfun_0.47 fs_1.6.4 sass_0.4.9 cli_3.6.3
[33] magrittr_2.0.3 grid_4.4.1 digest_0.6.37 rstudioapi_0.16.0
[37] askpass_1.2.0 lifecycle_1.0.4 vctrs_0.6.5 RSpectra_0.16-2
[41] evaluate_0.24.0 glue_1.7.0 whisker_0.4.1 cellranger_1.1.0
[45] fansi_1.0.6 rmarkdown_2.28 tools_4.4.1 pkgconfig_2.0.3
[49] htmltools_0.5.8.1